
April 2025
CASE HISTORY
A 16-year-old male presented with short stature, bilateral ptosis, diplopia, and Type 1 diabetes mellitus. Family history is significant for short stature and ptosis.
CASE CONTRIBUTED BY
Dr. Gauri Bornak, Radiology, NIMHANS, Bengaluru
Dr. Arka Prabha Chattopadhyay, Radiology, NIMHANS, Bengaluru
Dr. Sabha Ahmed, Radiology, NIMHANS, Bengaluru
Figure 1: Axial T2 (A and B) sequences of the brain show fairly symmetric areas of T2 hyperintensities involving the subcortical white matter (including subcortical U fibres) in bilateral frontal, parietal and temporal lobes (block arrows in A). There is sparing of the deep periventricular white matter. T2 hyperintense signal changes are also seen involving the bilateral globus pallidi, ventral thalami and the splenium of corpus callosum (line arrows in B). Axial FLAIR sequences (C and D) reveals hyperintense signal changes in the pontine tegmentum, bilateral superior cerebellar peduncles (curved arrow in C) and the dentate nuclei (curved arrow in D). Note is also made of diffuse generalized cerebral atrophy.
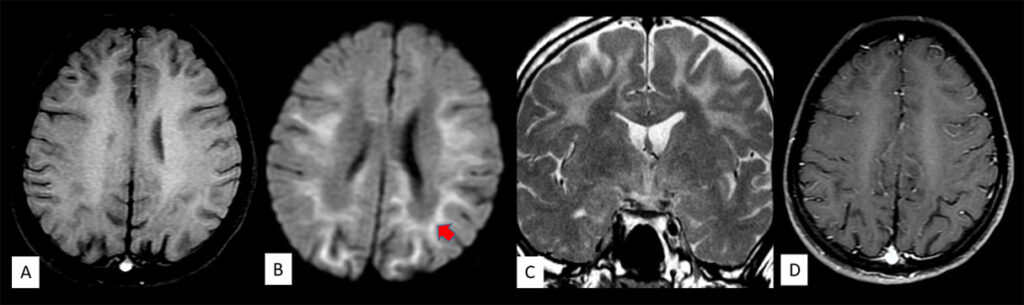
Figure 2: Axial T1 (A) sequence of the brain shows that the signal changes are isointense on T1. There is corresponding diffusion
restriction seen (block arrows in B). Coronal T2 (C) sequence also shows symmetric areas of T2 hyperintensities in bilateral subcortical white matter with sparing of the deep periventricular white matter. No corresponding contrast enhancement is seen.


Diagnosis: Kearns Sayre syndrome
Background: Kearns-Sayre syndrome (KSS) is a rare sporadic mitochondrial cytopathy caused by a single large-scale mitochondrial DNA (mtDNA) deletion. It is a clinical subtype of chronic progressive external ophthalmoplegia (CPEO). Classically, KSS has a triad of features including onset before 20 years of age, progressive external ophthalmoplegia (PEO), and pigmentary retinopathy.
Epidemiology: KSS is very rare. While the exact prevalence of this condition is unknown, one study has reported a prevalence of 1.6 cases per 100,000 in Western population.
Clinical presentation: Patients will have features of chronic progressive external ophthalmoplegia in the form of ptosis and paralysis of the extraocular muscles. Pigmentary retinopathy is another common feature. In addition, patients also present with cardiac conduction disturbances, non-ocular muscle weakness, SNHL, cerebellar ataxia and intellectual disability. The frequency of endocrine disturbances has been reported to range from 35% to 67%, including the following: diabetes mellitus, short stature and growth hormone insufficiency, hypogonadotropic hypogonadism, adrenal insufficiency, and primary hypoparathyroidism.
Pathology: Approximately 90% cases of KSS are sporadic, arising from a large-scale 1.1 to 10 kilobase deletion of mitochondrial DNA, with the most common deletion labeled as the “common 4977 bp deletion”, accounting for more than one-third of cases. In KSS muscle fibers stains demonstrate ragged red fibers like in other mitochondrial myopathies. Neurological findings of KSS are due to loss of myelin (however the exact mechanism is unknown).
Imaging features: The central nervous system (CNS) involvement is reflected by the extent of MRI abnormalities which can include T2/FLAIR hyperintensities involving predominantly the subcortical white matter, brain stem, globus pallidus, thalamus, and white matter of the cerebellum. Diffusion restriction can be seen. In addition, regional abnormalities of brain metabolism can be demonstrated in patients with KSS using MR spectroscopy (MRS). These could include an increase in the lactate (due to impairment of oxidative metabolism) as well as a significant decrease in N-acetylaspartate/creatine (a measure of neuron loss or dysfunction) in the cerebral cortex.
Differential diagnosis:
- L2 hydroxyglutaric aciduria: Patients present with complaints of developmental delay, seizures and macrocephaly. MRI shows centripetal pattern of involvement of white matter affecting the subcortical U fibres first, and then progressing to a deeper confluent pattern. The striatum and the dentate nuclei are commonly involved. There is sparing of the brainstem and the cerebellar white matter. Elevated levels of L-2 hydroxygluratic acid are seen in urine, plasma and CSF.
- TACO1 gene mutation: It manifests as Leigh phenotype. Clinical phenotype includes childhood-onset progressive cerebellar and pyramidal syndrome with optic atrophy and learning difficulties. MRI shows periventricular white matter lesions with multiple cystic/spongiform changes, involvement of the striatum, diencephalon (mainly the medial aspect of the thalamus), and brainstem (substantia nigra, oculomotor nuclei, periaqueductal gray matter, and inferior olivary nuclei).
Treatment: Management is supportive vigilance for detection of associated problems. Aponeurogenic ptosis and cricopharyngeal achalasia can be addressed surgically. Dietary and pharmacological agents include creatine monohydrate (CrM), α-lipoic acid, Vitamin C, E, and K (antioxidants), lutein, selenium (antioxidant), and coenzyme Q10. Hormonal replacement therapy in endocrinopathies and cardiac pacemaker placement for patients with cardiac conduction blocks.
References:
- Shemesh A, Margolin E. Kearns-Sayre Syndrome. [Updated 2023 Jul 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025
- Chu, B., Terae, S., Takahashi, C. et al. MRI of the brain in the Kearns- Sayre syndrome: report of four cases and a review. Neuroradiology 41, 759–764 (1999).
- Oktay Y, Güngör S, Zeltner L, Wiethoff S, Schöls L, Sonmezler E, Yilmaz E, Munro B, Bender B, Kernstock C, Kaemereit S, Liepelt I, Töpf A, Yis U, Laurie S, Yaramis A, Zuchner S, Hiz S, Lochmüller H, Schüle R, Horvath R. Confirmation of TACO1 as a Leigh Syndrome Disease Gene in Two Additional Families. J Neuromuscul Dis. 2020;7(3):301-308.
Add a Comment
You must be logged in to post a comment