
September 2025
CASE HISTORY
- A 2 -year-old female presented to the pediatrician with complaints of poor feeding and developmental delay
- History of Seizures (GTCS): 3 episodes in last 2 months
- Clinical Examination: Developmental Delay (Motor and Speech) and poor muscle tone.
- No history of similar complaint in sibling
- Non-Consanguineous marriage
CASE CONTRIBUTED BY
Dr. Manik Mahajan, Assistant Professor, Department of Radiology, Government Medical College, Jammu
Dr. Ramandeep Singh, Assistant Professor, Department of Radiology, Government Medical College, Jammu
Figures A-H:
Axial T2 (A,E) and FLAIR (B) images showing symmetrical T2 (Black arrow) and FLAIR (White arrow) hyperintensity involving the cerebellar and cerebral white matter (periventricular, deep and subcortical white matter), medulla, pons and mid brain, lateral thalami, globus pallidi and perirolandic region. Axial DWI (C,G) and corresponding ADC (D,H) maps show marked DWI hyperintensity (Orange arrows) and ADC hypointensity (Red arrows) in the involved areas representing intramyelinic edema. On T1 (F) images, diffuse hypointensity (Blue arrow) is seen in the involved cerebral white matter.
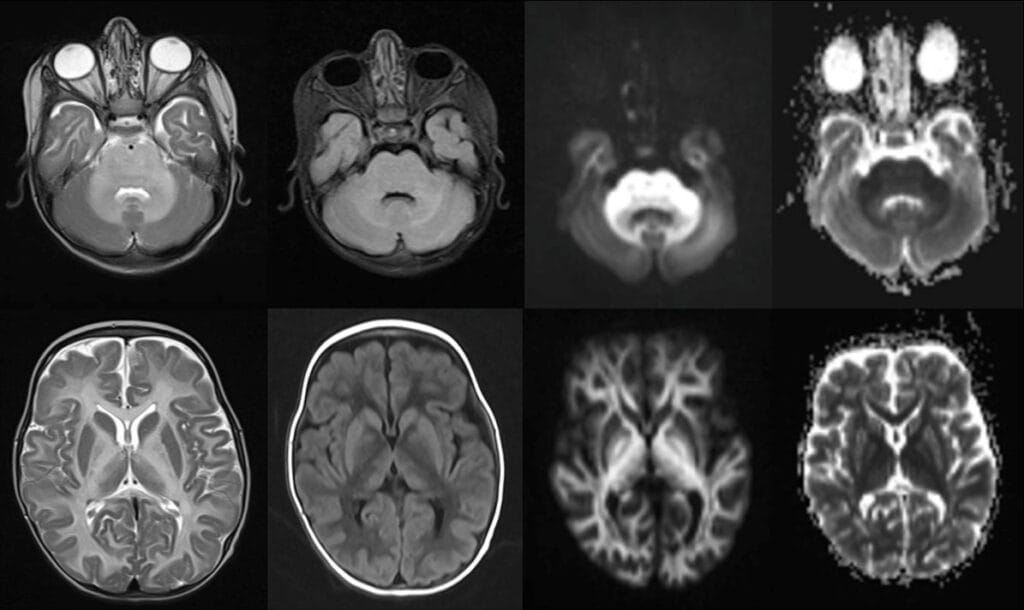
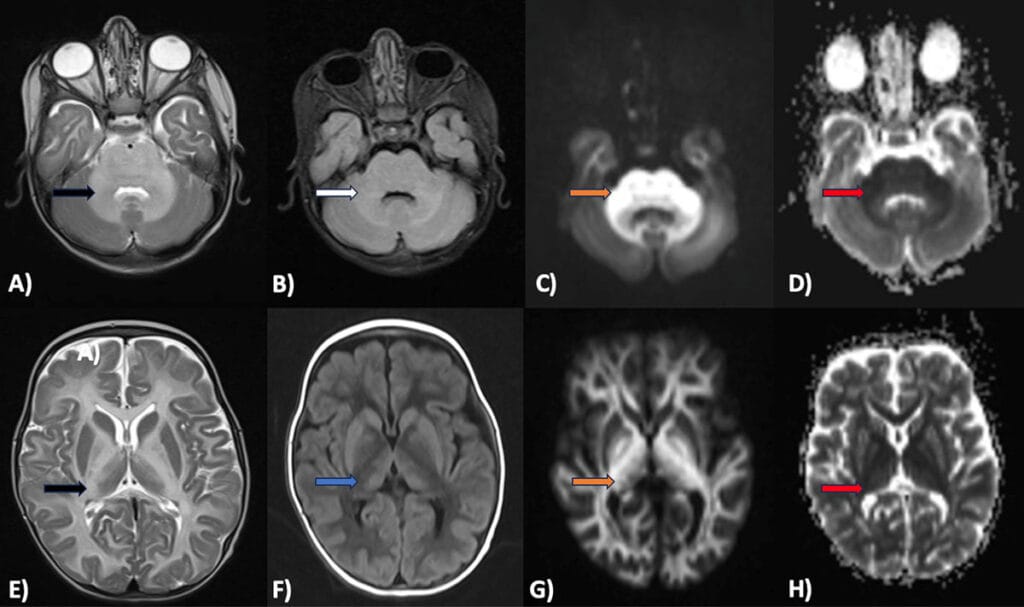
Diagnosis: Maple Syrup Urine Disease (MSUD)
Background:
- Maple syrup urine disease (MSUD) is a rare autosomal-recessive (AR) disorder
- Caused by deficiency of branched-chain alpha-keto acid dehydrogenase enzyme [1].
- MSUD is very rare with an an estimated worldwide incidence of just 1 case per 185,000 live births [2].
Pathophysiology:
- MSUD is caused by an AR mutation in 1 of the 3 genes: BCKDHA, BCKDHB, or DBT.
- Accumulated BCAA and alpha-ketoacids lead to a constellation of clinical symptoms due to CNS, immune system, and skeletal muscle dysfunction [3].
- High intracranial leucine competes with the cerebral uptake of several other important amino acids, particularly phenylalanine, glutamine, histidine, methionine, and tryptophan, negatively impacting brain growth, neurotransmitter production, and myelin synthesis and cognitive dysfunctions [4].
- This results in decreased blood osmolarity, low sodium concentrations, and increased intracellular water, leading to cerebral edema [5].
Clinical presentation:
- MSUD is classified into classic, intermediate, intermittent, E3-deficient, and thiamine-responsive.
- Individuals with the classic neonatal type exhibit a maple syrup odor in the cerumen and urine and develop neurological features such as poor feeding, irritability, apnea, opisthotonos, and “bicycling” movements. Cerebral edema is mostly present, leading to coma and early death.
- Infants with the intermediate form of the disease typically present later in the first years of their lives with complaints of feeding problems and intellectual disability.
- Infants diagnosed with intermittent forms show normal growth and neurological development without any symptom onset, even if kept on an unrestricted diet. However, during catabolic situations, these patients also exhibit the clinical and biochemical characteristics of the classic form
- The thiamine-responsive form has the best prognosis compared to other affected individuals with classic and intermittent forms of the disease
Key Imaging Findings:
- MRI plays a crucial role in the diagnosis.
- MSUD patients have a characteristic pattern of brain edema, particularly during the MSUD crisis.
- Two forms of edema are seen, namely, intramyelinic edema and vasogenic edema with the former being more common cause in classical cases.
- Characteristic brain regions involved include cerebellar white matter, peduncles, dorsal brainstem, peri-rolandic cerebral white matter, thalami, globus palladi, and the posterior limb of the internal capsule.
- Involved regions appear hypointense on T1-weighted imaging and hyperintense on T2-weighted imaging.
- Regions with diffusion restriction are particularly prominent in the posterior limb of the internal capsule and corticospinal tract.
Differential Diagnosis:
- HIE
- Non Ketotic Hyperglycinemia
- Urea Cycle Disorders
- Pyruvate Dehydrogenase Complex Deficieny
Management and Prevention:
- Early treatment is essential to prevent onset of neurological side effects.
- Most patients survive and do not develop neurological effects if treated within a few days. However, once neurological symptoms appear, the damage is usually permanent and irreversible.
- Dietary management includes a balance between providing normal growth and development to the patient by giving enough protein and BCAAs and trying to ensure that the patient’s condition and BCAA levels are within normal limits.
- A chorionic villus biopsy or amniocentesis may be done in patients with a family history of MSUD to identify the disorder before birth
References:
- K K H, Ajmera P, Agarwal A, Dahiya A, Parripati VK. Maple Syrup Urine Disease: An Uncommon Cause of Neonatal Febrile Seizures. Cureus. 2023 Jun 22;15(6):e40826.
- Edelmann L, Wasserstein MP, Kornreich R, Sansaricq C, Snyderman SE, Diaz GA. Maple syrup urine disease: identification and carrier-frequency determination of a novel founder mutation in the Ashkenazi Jewish population. Am J Hum Genet. 2001 Oct;69(4):863-8.
- Hassan SA, Gupta V. Maple Syrup Urine Disease. 2024 Mar 3. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan
- Strauss KA, Puffenberger EG, Carson VJ. Maple Syrup Urine Disease. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. University of Washington, Seattle; Seattle (WA): Jan 30, 2006.
- Morton DH, Strauss KA, Robinson DL, Puffenberger EG, Kelley RI. Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics. 2002 Jun;109(6):999-1008.
Add a Comment
You must be logged in to post a comment