
July 2025
CASE HISTORY
A 22-year-old male, born of nonconsanguineous parentage, presented with insidious onset, gradually progressive symmetrical upper limb and lower limb ataxia, dysarthria, and distal upper and lower limb weakness since the age of eight years. Saccadic intrusions were present, characterized by slow saccades and broken pursuits. His sibling and two maternal uncles were affected with similar complaints. Nerve conduction study showed bilateral symmetrical demyelinating polyneuropathy.
CASE CONTRIBUTED BY
Dr. Rakesh, Senior Resident
Dr. Smily Sharma, Assistant Professor
Dr Bejoy Thomas, Senior Grade Professor
Dr C Kesavadas, Senior Grade Professor and Head of the Department
Affiliation: Department of Imaging Sciences and Interventional Radiology, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Trivandrum, Kerala, India
Imaging Findings
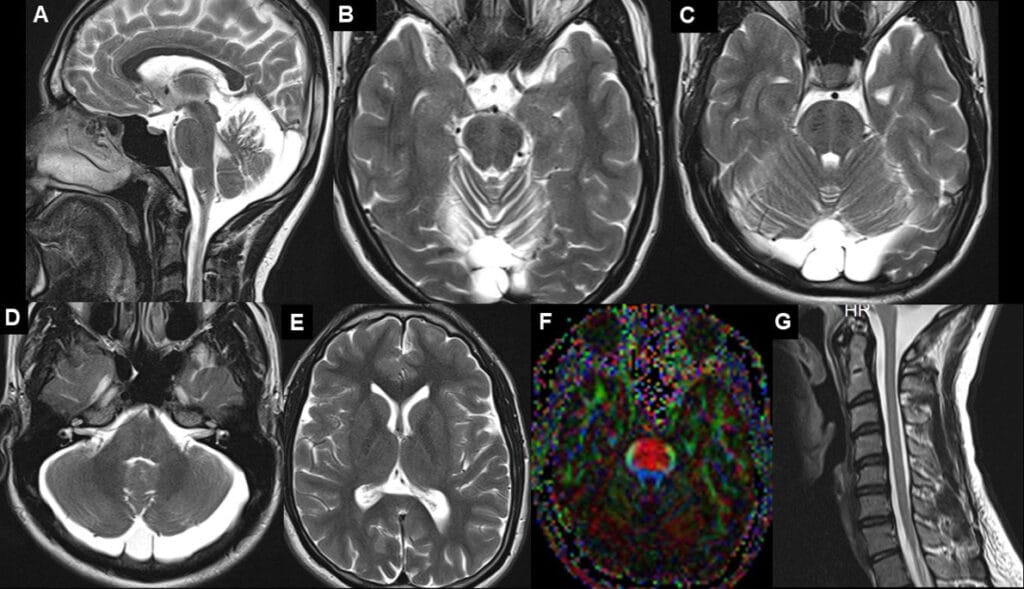
Sagittal T2-weighted image shows enlarged pons (broken arrows) with a significant superior vermian atrophy (red arrow). Mild thinning of the posterior body of the corpus callosum is seen (yellow solid arrow). B to D. Characteristic transverse T2 hypointense linear striations are seen in the pons on either side of the midline (yellow arrows). Additional T2 hyperintensities are seen in the lateral pons at the level of the middle cerebellar peduncle (red broken arrows). Note the superior vermian atrophy (red arrow in B) and prominent retrocerebellar CSF space (orange solid arrow in D). E. Mild supratentorial volume loss is also seen with mildly prominent sulcal spaces. F. DTI image shows increased thickness of the transverse pontine fibres (yellow arrows: red colour) with thinning of the descending corticospinal tract fibers. F. Spine screening did not show significant cord atrophy or hyperintensities. Overall, imaging features are characteristic for Autosomal Recessive Spastic Ataxia Of Charlevoix Saguenay (ARSACS).

Whole exome and mitochondrial genome sequencing was done. SACS-Exon 10, c8793 del, homozygous, autosomal recessive, pathogenic variant was found.
Diagnosis: AUTOSOMAL RECESSIVE SPASTIC ATAXIA OF CHARLEVOIX SAGUENAY (ARSACS)
Clinical presentations:
ARSACS is a neurodegenerative disorder that is characterized by cerebellar ataxia, pyramidal tract signs, and peripheral neuropathy. Patients present with spasticity, gait disturbances, and walking difficulties at a young age (12–18 months), which progress throughout life. They are usually wheelchair bound by the fifth decade. Axonal polyneuropathy usually manifests by the third decade. Non-progressive ocular disturbances, including saccadic alteration of ocular pursuit and retinal hypermyelination (without vision loss), may also be present early in the disease. Other described associations include mitral valve prolapse, pes cavus, and bladder dysfunction.
Genetics:
It occurs due to autosomal recessive mutations in SACS gene, located on chromosome 13q. The SACS gene encodes the sacsin protein, which is expressed in the brain, skin, skeletal muscles, and minimally in the pancreas.
Key imaging features :
- Enlarged pons with linear T2/ FLAIR hypointense transverse striations (tigroid pattern/ striped appearance) on either side of the midline. These likely represent abnormally large transverse pontocerebellar fibers (also depicted on DTI).
- Superior cerebellar vermian atrophy, which occurs early in the disease suggesting its congenital nature
- Other less specific findings include:
- T2 hyperintense rim around the thalami
- Cervical spinal cord atrophy
- Thinning of the corpus callosum
- Progressive Cerebellar atrophy
- Cerebral atrophy in later life
- Treatment is mainly supportive
Differential diagnosis:
- Friedreich ataxia: typically present with decreased AP diameter of the medulla oblongata and cervical spinal cord combined with symmetric T2 hyperintensity in the lateral or dorsal columns of the spinal cord; relative preservation of the cerebellar volume or mild upper vermian atrophy. Lack of characteristic T2 hypointense striations in pons.
- Spinocerebellar ataxia type 3 (SCA3): MRI findings include atrophy of the vermis, cerebellar hemispheres, and middle cerebellar peduncles, and in the later stages can show linear hyperintensities in the midline of the pons coursing in an anteroposterior direction.
- Ataxia with vitamin E deficiency (AVED): MRI is typically normal or may show mild cerebellar and vermian atrophy.
References
- Divya KP, Cherian A, Dhing HK, Kumar S, Thomas B, Faruq M. Widening the clinical, radiological and genetic spectrum of autosomal recessive ataxia of Charlevoix-Saguenay in Indian patients. Acta Neurologica Belgica. 2024 Apr;124(2):475-84.
- Martin MH, Bouchard JP, Sylvain M, St-Onge O, Truchon S. Autosomal recessive spastic ataxia of Charlevoix-Saguenay: a report of MR imaging in 5 patients. American journal of neuroradiology. 2007 Sep 1;28(8):1606-8.
- Biswas A, Varman M, Yoganathan S, Subhash PK, Mani S. Teaching NeuroImages: autosomal recessive spastic ataxia of Charlevoix-Saguenay: Typical MRI findings. Neurology. 2018 Apr 3;90(14):e1271-2.
- Karuvath RH, Patwari S, Chadaga H. Case 293: Autosomal recessive spastic ataxia of Charlevoix-Saguenay. Radiology. 2021 Sep;300(3):730-2.
Add a Comment
You must be logged in to post a comment